Recombinant Human Probable phospholipid-transporting ATPase IC (ATP8B1), partial
-
中文名稱:Recombinant Human Probable phospholipid-transporting ATPase IC(ATP8B1) ,partial
-
貨號:CSB-YP002418HU
-
說明書:
-
規格:
-
來源:Yeast
-
其他:
-
中文名稱:Recombinant Human Probable phospholipid-transporting ATPase IC(ATP8B1) ,partial
-
貨號:CSB-EP002418HU
-
說明書:
-
規格:
-
來源:E.coli
-
其他:
-
中文名稱:Recombinant Human Probable phospholipid-transporting ATPase IC(ATP8B1) ,partial
-
貨號:CSB-EP002418HU-B
-
說明書:
-
規格:
-
來源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名稱:Recombinant Human Probable phospholipid-transporting ATPase IC(ATP8B1) ,partial
-
貨號:CSB-BP002418HU
-
說明書:
-
規格:
-
來源:Baculovirus
-
其他:
產品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:ATP8B1
-
Uniprot No.:
-
別名:AT8B1_HUMAN; ATP8B1; ATPase class I type 8B member 1; ATPase; aminophospholipid transporter; class I; type 8B; member 1; ATPIC; BRIC; E1-E2 ATPase; Familial intrahepatic cholestasis type 1; FIC1; OTTHUMP00000163615; PFIC; PFIC1; Phospholipid transporting ATPase IC; Probable phospholipid-transporting ATPase IC
-
種屬:Homo sapiens (Human)
-
蛋白長度:Partial
-
蛋白標簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:Catalytic component of a P4-ATPase flippase complex which catalyzes the hydrolysis of ATP coupled to the transport of phospholipids, in particular phosphatidylcholines (PC), from the outer to the inner leaflet of the plasma membrane. May participate in the establishment of the canalicular membrane integrity by ensuring asymmetric distribution of phospholipids in the canicular membrane. Thus may have a role in the regulation of bile acids transport into the canaliculus, uptake of bile acids from intestinal contents into intestinal mucosa or both and protect hepatocytes from bile salts. Involved in the microvillus formation in polarized epithelial cells; the function seems to be independent from its flippase activity. Participates in correct apical membrane localization of CDC42, CFTR and SLC10A2. Enables CDC42 clustering at the apical membrane during enterocyte polarization through the interaction between CDC42 polybasic region and negatively charged membrane lipids provided by ATP8B1. Together with TMEM30A is involved in uptake of the synthetic drug alkylphospholipid perifosine. Required for the preservation of cochlear hair cells in the inner ear. May act as cardiolipin transporter during inflammatory injury.
-
基因功能參考文獻:
- FIC1, BSEP, and MDR3 represent hepatobiliary transport proteins essential for bile formation. PMID: 28733223
- ATP8B1 deficiency caused incomplete polarization of human peripheral blood monocyte-derived macrophages into M2c, a subset alternatively activated macrophages. PMID: 29104077
- Patients with a confirmed ABCB11 or tight junction protein 2 gene mutation (n = 7) had a minimally detectable THBA proportion (0.23-2.99% of total BAs). Three patients with an ATP8B1 mutation had an elevated THBA proportion (7.51-37.26%). PMID: 28073941
- the first characterisation at the protein level of six ABCB4 variants (D243A, K435T, G535D, I490T, R545C, and S978P) previously found in patients with inflammatory liver diseases or liver cancer, is reported. PMID: 28220208
- The lipid flippases, ATP8B1 and ATP11A are novel elements of the innate immune response that are essential to attenuate the inflammatory response, possibly by mediating endotoxin-induced internalization of TLR4. PMID: 27628304
- ATP8B1 is important for proper CFTR expression and function. PMID: 27301931
- GGT levels in patients with ATP8B1 or ABCB11 deficiency varied with age. The peak GGT value was <70U/L in the 2nd~6th month of life, <60U/L in the 7th~12th month and <50U/L beyond one year PMID: 27050426
- As hypothyroidism can be another extrahepatic feature of ATP8B1 deficiency, thyroid function should be monitored in these patients. PMID: 26382629
- the predominant P4 ATPases in pure pancreatic beta cells and human and rat pancreatic islets were ATP8B1, ATP8B2, and ATP9A. ATP8B1 and CDC50A were highly concentrated in ISG PMID: 26240149
- insufficient activity of Atp8b1/FIC1 increases susceptibility to bacterial pneumonia. PMID: 26050466
- We systematically characterized the molecular consequences of 14 ATP8B1 mutations at exon-intron boundaries associated with ATP8B1 deficiency and found that the majority resulted in total exon skipping PMID: 25421123
- Data indicate that the lipid flippase (ATP8B1)-transmembrane protein 30A (CDC50A) heterodimer is essential for the apical localization of sodium-dependent bile acid transporter (SLC10A2/ASBT) in Caco-2 cells. PMID: 25239307
- We did not find an association between heterozygous ATP8B1 variants and chronic pancreatitis in our cohort of patients with hereditary and idiopathic chronic pancreatitis. PMID: 24260417
- Case Report: suggest contribution of ATP8B1 mutations to drug-induced liver injury from anabolic androgenic steroids marketed as dietary supplements. PMID: 23750872
- Case Report: missense ATP8B1 mutation in adult male with progressive familial intrahepatic cholestasis type 1. PMID: 23197899
- Novel splice-site mutation in ATP8B1 results in atypical progressive familial intrahepatic cholestasis type 1. PMID: 23033845
- FIC1 signals to FXR via a signaling pathway including PLD2 and PKCzeta PMID: 23213138
- The basal expression of ATP8B1 is driven by a housekeeping-like promoter located 71 kb upstream of the first protein coding exon, and is independent of bile acids and farnesoid X receptor. PMID: 23251605
- Biochemical analysis of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis PMID: 23060447
- Sequence analysis of the genes identified 27% cholestasis subjects with missense, nonsense, deletion, and splice site variants associated with disease phenotypes based on the type of mutation in the JAG1, ATP8B1, ABCB11, or ABCB4 genes. PMID: 20683201
- results unveil a new paradigm whereby Atp8b1 is a cardiolipin importer whose capacity to remove cardiolipin from lung fluid is exceeded during inflammation or when Atp8b1 is defective PMID: 20852622
- facilitating diagnosis and elucidating the differing consequences of ATP8B1 and ABCB11 mutations in progressive familial intrahepatic cholestasis PMID: 20447715
- Data identified three novel mutations in BSEP, one novel mutation in MDR3, and one heterozygous mutation in ATP8B1 in PFIC patients. PMID: 20414253
- We now report evidence that heterozygous genetically determined alteration of ATP8B1 (encoding FIC1) may also represent a risk factor for transient neonatal cholestasis. PMID: 20216097
- critical role in apical membrane organization that is unrelated to its presumed aminophospholipid translocase activity PMID: 20512993
- ATP8B1 gene mutations play an important role in Chinese patients with progressive intrahepatic cholestasis and low gamma-glutamyltransferase. The linked mutation P209T and IVS6+5G>T is a hot mutation in the Chinese population. PMID: 20038848
- Progressive familial intrahepatic cholestasis types 1 & 2 differ clinically, biochemically, and histologically at presentation and/or during the disease course. A small proportion of normal-GGT PFIC is likely not due to ATP8B1 or ABCB11 mutations. PMID: 20232290
- Findings support the hypothesis that hepatocyte FIC1 enhances FXR signaling via a PKCzeta-dependent signaling pathway. PMID: 19809379
- A surprisingly large proportion of ATP8B1 mutations resulted in aberrant folding and decreased expression at the plasma membrane. These effects were partially restored by treatment with 4-phenylbutyrate. PMID: 19918981
- three homologues found and two sequenced and two RNA transcript sizes analysed by northern blot: APT8B2, APT8B3, APT8B4 PMID: 12880872
- Loss of familial intrahepatic cholestasis-1 leads to diminished nuclear translocation of farnesoid X receptor(FXR), with subsequent potential for pathologic alterations in intestinal and hepatic bile acid transporter expression PMID: 14988830
- 54 distinct disease mutations: 10 mutations predicted to disrupt splicing, 6 nonsense mutations, 11 small insertion or deletion mutations predicted to induce frameshifts, 1 large genomic deletion, 2 small inframe deletions, and 24 missense mutations. PMID: 15239083
- Coexpression with CDC50 proteins resulted in relocalization of ATP8B1 from the endoplasmic reticulum to the plasma membrane. In the plasma membrane, ATP8B1 functions as a flippase for phosphatidylserine. PMID: 17948906
- FIC1 signals to FXR via PKC zeta. FIC1-related liver disease is likely related to downstream effects of FXR on bile acid homeostasis. PMID: 18668687
- ATP8B1 deficiency predisposes to cholestasis by favoring bile acid-induced injury in the canalicular membrane but does not directly affect FXR expression PMID: 19027009
- Knockdown of ATP8B1 expression leads to specific downregulation of the bile acid sensor FXR in HepG2 cells: effect of the FXR agonist GW4064. PMID: 19228886
- These results suggest that the PFIC1 mutants have a lower stimulatory effect on FXR activity and cannot interact with CDC50A, which may lead to the development of the features of PFIC1. PMID: 19381753
- show that ATP8B1/Atp8b1 deficiency, both in patients and in Atp8b1(G308V/G308V) mutant mice, causes hearing loss, associated with progressive degeneration of cochlear hair cells PMID: 19478059
- Post-liver transplantation steatosis may be due to a malfunction of the ATP8B1 product. PMID: 19479804
- PFIC1 mutations lead to the complete absence of canalicular expression, whereas in BRIC1/ICP residual protein is expressed in the canalicular membrane. PMID: 19731236
顯示更多
收起更多
-
相關疾病:Cholestasis, progressive familial intrahepatic, 1 (PFIC1); Cholestasis, benign recurrent intrahepatic, 1 (BRIC1); Cholestasis of pregnancy, intrahepatic 1 (ICP1)
-
亞細胞定位:Cell membrane; Multi-pass membrane protein. Apical cell membrane. Cell projection, stereocilium. Endoplasmic reticulum. Golgi apparatus.
-
蛋白家族:Cation transport ATPase (P-type) (TC 3.A.3) family, Type IV subfamily
-
組織特異性:Found in most tissues except brain and skeletal muscle. Most abundant in pancreas and small intestine.
-
數據庫鏈接:
Most popular with customers
-
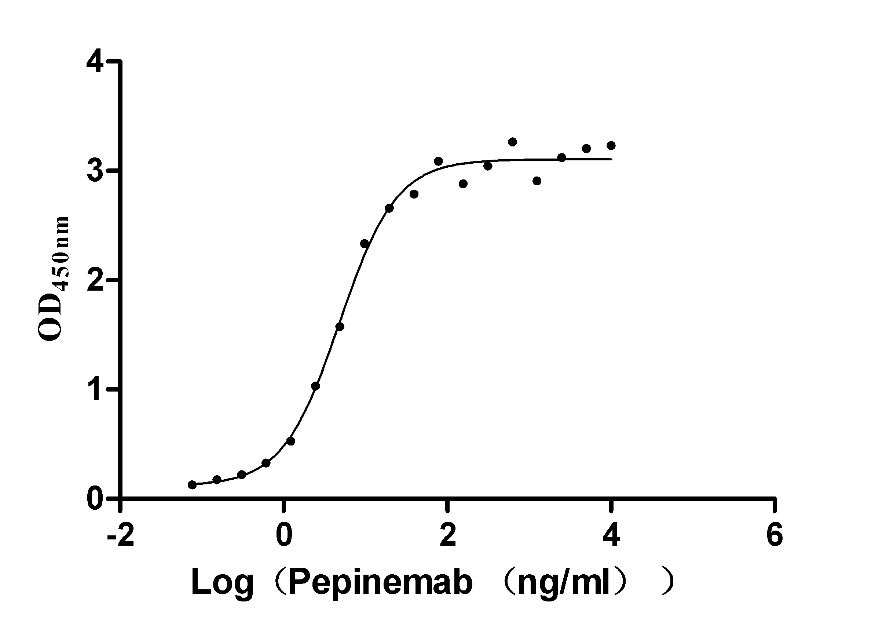
Recombinant Human Semaphorin-4D (SEMA4D), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Human Insulin growth factor-like family member 1 (IGFL1) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
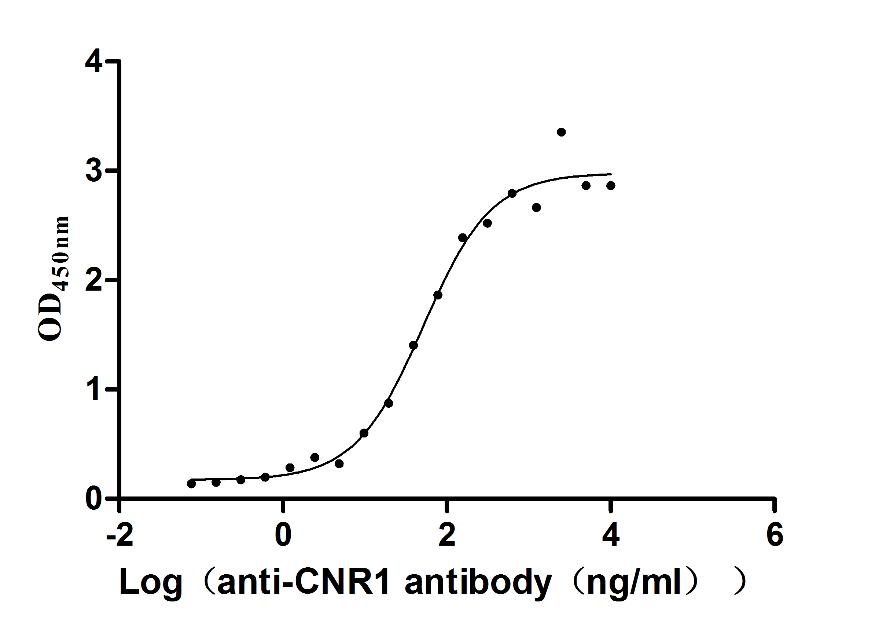
Recombinant Human Cannabinoid receptor 1 (CNR1)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
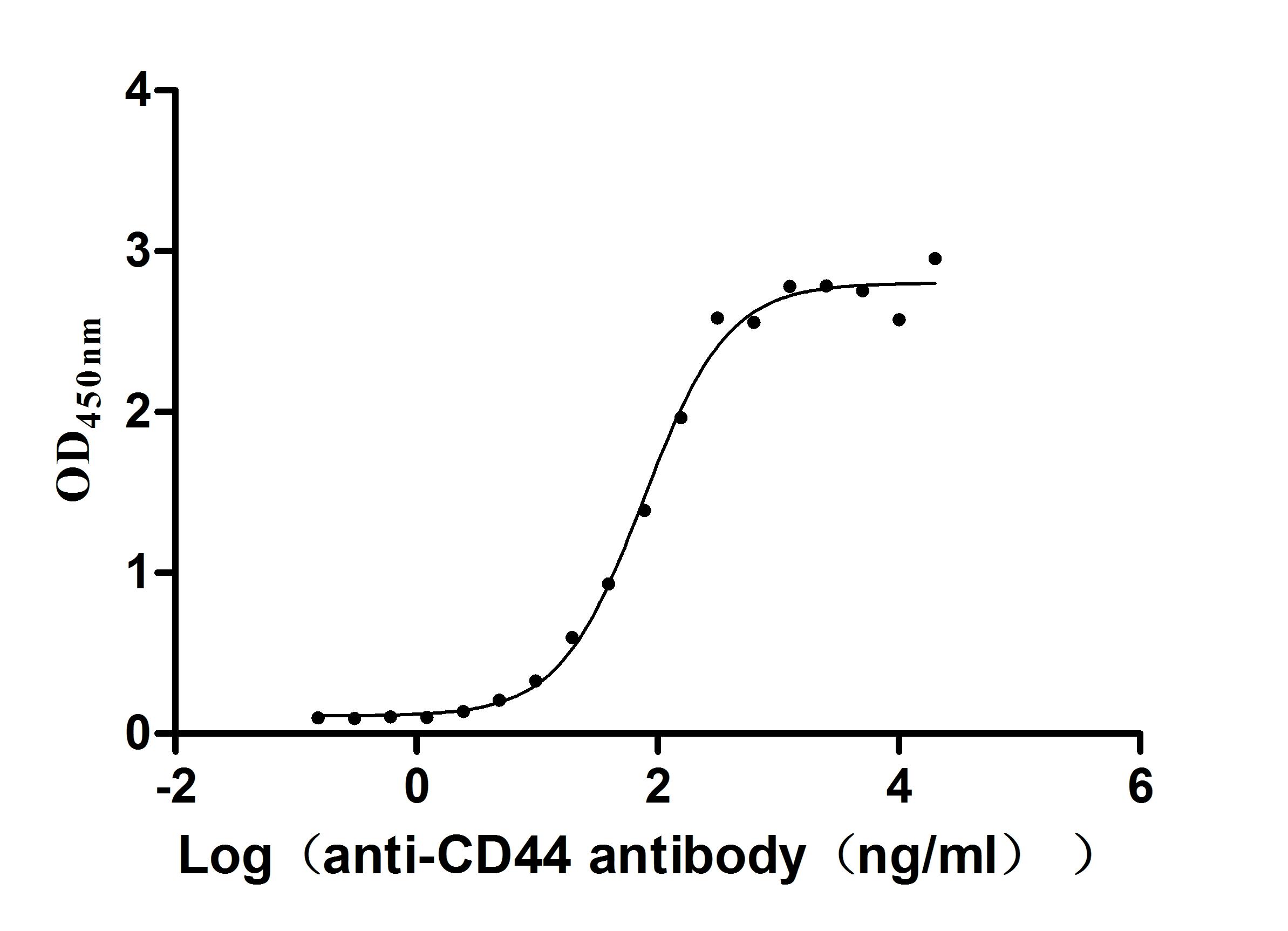
Recombinant Macaca fascicularis CD44 antigen (CD44), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
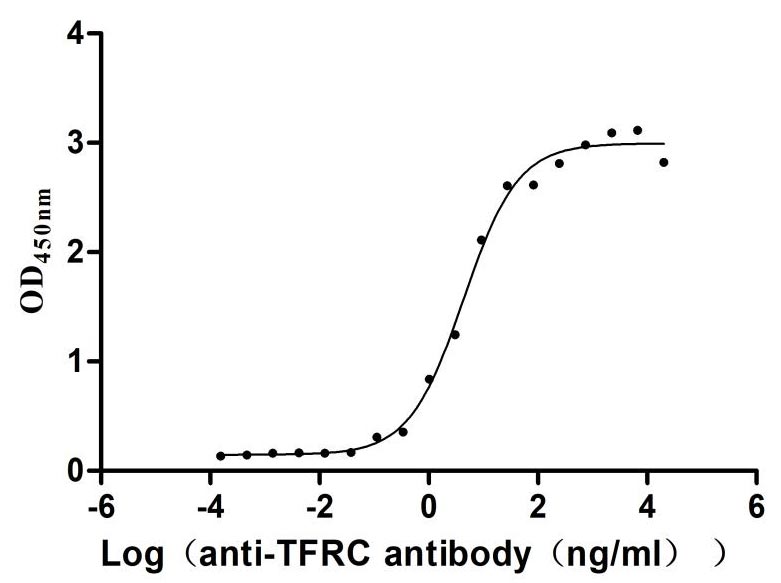
Recombinant Human Transferrin receptor protein 1 (TFRC), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Human CD81 antigen (CD81), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Macaca fascicularis C-type lectin domain family 4 member C(CLEC4C), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Human C-C chemokine receptor type 9 (CCR9)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)









